

Cognitive and emotional disorders in early stages of Parkinson's disease: in search of new genetic models
2014/01/21
Abstract
Since a long time it has been shown that Parkinson's Disease does not only affect motor system, but also cognitive and emotional circuits. These non-motor symptoms appear in early stages,and could be a way to diagnose the disease before a too serious degeneration. It is why many tests to realize have been performed on humans with different questions leading to observations of emotional troubles, sleep disorders, depression, apathy. But the research is difficult because of the hard task to generate models exhibiting these non-motor symptoms. Indeed, genetic models (α-synuclein, Parkin or LRRK2) develop motor impairments easier than cognitive and emotional ones. However some of them develop symptoms like dementia or psychiatric impairments giving hope to find the right model.
Table of Contents
Introduction
Detecting Parkinson's Disease (PD) in early stages is becoming a priority. Indeed, when first motor symptoms (MS) like tremor, difficulties to start a movement, etc. appear, treatments are not efficient anymore: 60% to 70% of neurons of the Substantia Nigra (SN) have already degenerated [1]. It is why researchers have to find a way to diagnose a patient with PD before this point of “non-return”. However, some non-motor symptoms (NMS) have been exhibited in PD patients by James Parkinson in 1817, such as sleep disorders and delirium which seemed to appear before MS, so in earlier stages. Then, many others NMS have been diagnosed and characterized at earliest stages of the neurodegenerative disease. Depression [2]•, apathy [3], alexithymia [4], sleep disturbances [5], hallucinations [6] and other cognitive troubles are often found in early PD patients. However, these NMS are common with others disorders which do not necessarily lead to development of PD. So it is essential to link these emotional and cognitive disturbances with anatomic and physiological lesions specific to PD. To establish this correlation, the NMS have to be scrupulously studied and rated in order to identify PD patients before the critical moment of “no return”. So, scales of measure need improvements and new mice models should be generated. Indeed it is very difficult to obtain a model which replicates most of the symptoms, motor and non-motor, and with histological and anatomical phenotypes corresponding to what is observed in humans PD patients. But it is not enough to mimic the symptoms obtained in PD patients: it would be more interesting if the cause of the disease leading to the establishment of the model was the same than the ones found in PD patients.
Cognitive and emotional disorders in PD patients in early stages
Depression
36% to 53% of European PD patients suffer from depression [7]. This NMS is really controversial because it is difficult to say if it is a consequence of the awareness of the disease (hard to realize that we are affected by an incurable disease) or whether it comes from degenerescence in the brainstem [8]•. It may be a mix of both. Moreover, depression shows different features such as suicide attempts, mood disorders, or weariness in many situations [9]. The Dorsal Raphe Nucleus (RD) seems to be involved in depression in PD with a reduction of neurons quantity and the appearance of Lewy Bodies (LB), which consist of α‑synuclein (α-syn) aggregates [10]. Many neurons of the RD are serotonergic (5-HT). But depression is also associated with depletion of others monoaminergic systems like dopaminergic (DA) and noradrenergic (NA) system found in the SN. Degeneration of neurons from the Locus Coeruleus (LC) precedes dopamine neurons degeneration within SN (Figure 1). Indeed, the decrease of DA and NA transporters from LC may be linked to depression [11].
Regarding the molecular and genetic pathway, Kasten and colleagues reviewed that PD patients who carries α-syn mutations developed depression: 39% of patients carrying A53T mutation, 40% with E46K mutation, 59% with a duplication of the gene, and all patients who present a triplication of it [12]. So there seems to be a link between the mutation and the severity of the disease.
Depressive symptoms can be evaluated with different tests or questionnaires. The Hamilton Rating Scale for depression (HAMD-A) measures the degree of severity of depression. Another method to assess mood is the Beck Depression Inventory (BDI). Developed by Aaron T. Beck in 1961, he revised it in 1978 (BDI1-A) and in 1996 (BDI-II). It consists in a 21 multiple choice questions about patients feelings in the last few days. The different possible answers are: 0 “I do not feel sad”, 1 “I feel sad”, 2 “I am sad all the time”, 3 “I am so sad or unhappy that I can't stand it”. The final score varies from 0 (minimal severity) to 63 (worse severity). It is interesting to notice that BDI-II is positively correlated with the HAMD-A with a Pearson R of 0.71 which valid both test [13]. Older scales as the Geriatric Depression Scale-15 (GDS-15) can be used to evaluate depression.
Apathy
Apathy is characterized by a decrease of motivation, with a reduced wish of participating to different activities and a weak consciousness. The patient also displays cognitive impairments or emotional distress, and other features: hypomimia, a reduction of facial mimics caused by motor and psychological factors, rigidity, axial impairment. It is explained by the impairment of the frontal lobes caused by damages of the frontal cortex. Frontal-subcortical circuits could be also involved leading to basal ganglia disorders (Figure 1) [14]•. Then, apathy can be evaluated with the Lille Apathy Rating Scale (LARS) which is a structured interview consisting in 33 items divided into 9 domains. This scale makes the discrimination possible between apathy and depression. It also organizes the trouble in four categories. So it allows researchers to assess that 16,5% to 42% of PD patients are apathetic. It has also been shown that apathy is correlated to loss of recall and executive functions [3]. The Wisconsin Card Sorting Test (WCST) is a way to measure it and to detect frontal lobe disturbances. That is why many researchers consider that apathy should be taken in consideration as an early sign of dysexecutive symptom in PD revealing troubles of cognitive processing.
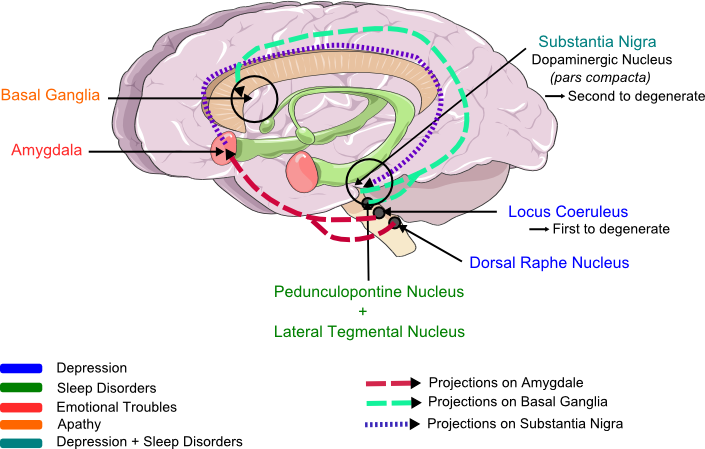
Figure 1. Structures involved in non-motor symptoms of Parkinson's Disease. .
Recapitulation of brain lesions correlated with the different NMS: depression (blue), apathy (orange), emotion (pink), sleep disorder (green). Degeneration of dopaminergic neurons are involved in all symptoms, but especially in depression. These different part of the brain are linked and explain the dependance between NMS. The different arrows illustrate the projections between nucleus and show the convergence of structures on the amygdala, which projects itself on Substantia Nigria and which is the emotion and feeling center. So this picture shows all the links explaining the strong correlation between the LC, the RD, the SN, the amygdala and the basal ganglia (the drawn way are not representative of the real circuit of the neurons projections). (Modified from Servier Medical Art.)
Emotions
PD patients suffer from abnormal recognition of facial emotion [20–22]. Bowers and colleagues showed that PD patients exhibit a "blunted startle eye-blink response" viewing nasty images [18] while Miller and colleagues observed that PD patients seem to be under-reactive when they are faced with an emotional stimulus [19]. Physiopathologically speaking, the disturbance may be coming from a pupil dilatation issue which is the consequence of sympathetic activation during the presentation of pictures involving emotional responses. It is good to know that the sympathetic way is regulated by the noradrenergic brain stem nuclei and inhibited by the parasympathetic way [20]. However, reactions of PD patients are different. Indeed, many studies tried to measure pupil dilatation diameter and ocular movements as an emotional processing to compare PD patients and controls viewing affective scenes. But results are really divergent. Some studies reveal a normal behavior for voluntary saccade movements but not for reflex [21] while Briand and colleagues show the opposite result [22]. Recently, another study shows results from hyper-reflexive saccades during emotional stimuli to a total disregard for theses emotional stimuli [23]. To conclude about the ocular processing, we can only say that when PD patients present emotional abnormalities, it might come from an amygdala atrophy (Figure 1) and the characteristic Lewy body deposition [18] even if no significant correlation between eye movements and PD severity has been shown.
Sleep Disorders
Demonstrations of sleep disorders arise years before MS [24]. 60% to 98% of PD patients suffer from it. Two phenomena are observed: the Rapid Eye Movement sleep Behavior Disorder (RBD) and the Excessive Day Sleepiness (EDS) [8].
RBD leads to a loss of normal skeletal muscle atonia due to the absence of motor activity. It is linked to a complex dysfunction of GABAergic, glutamatergic and cholinergic systems due to a degeneration of brainstem nuclei in the latero-dorsal pontine tegmentum. Indeed, in normal REM-sleep, cholinergic neurons are strongly active, leading to the hypothesis of a disruption in the processing of cholinergic facilitation for REM-sleep. For EDS, other structures impairments seem to be involved such as pedunculopontine nucleus (PPN), the lateral tegmental nucleus (LTN), the LC [8]•[24]••, the DR [8] and their anatomical connections (amygdala, pallidum, neocortex...) (Figure1) [24]••. But the direct link between sleep disturbances and theses structures remains unclear [8]. Several scales are used to diagnose sleep troubles in PD patients: the Pittsburgh Sleep Quality Index (PSQI) assesses the nocturnal sleep impairment while the Epworth Sleepiness Scale (ESS) estimates daytime somnolence (significant when score is higher than 10 because the maximum score corresponding to the strongest difficulties is 21).
A Global Study
In order to evaluate NMS of PD in a global way and to have the possibility to rate severity or disease stage, a first questionnaire has been developed as a primary screening instrument for NMS. It is the Non-Motor Symptoms Questionnaire (NMSQuest) evaluating cognitive and sleep disorders with "present" or "absent" responses (Figure 2) [25]••. In addition, the Unified Parkinson's Disease Rating Scale (UPDRS) evaluates the severity of the disease for patients that have already been diagnosed as affected by PD. It is used in studies in order to make homogenous patients groups (same stage in the disease) and performed only at the third part of the study because it tests motor activities: it shows the important place that NMS takes in the disease[26]. Duncan and colleagues recently made a global study with an array of questionnaires: the NMSQuest, the Mini-Mental State (MMSE) and the Montral Cognitive Assessment (MoCA) evaluating global cognitive functions, the GDS-15, the PSQI and the ESS. Thanks to these tests, authors observed different NMS such as attention, apathy, memory, hallucinations, disillusions, depression and anxiety, sleep. It was demonstrated that PD patients suffer more from NMS than controls: mean number 8.3 ± 4.3 symptoms for PD patients versus 2.8 ± 2.5 for controls (P<0.001). Many PD patients displayed memory troubles (53.8%) but many controls complained of the same cognitive impairment (40.8%). Moreover, MMSE and MoCA showed significant reduced scores in PD patients compared to controls and increases of depression and anxiety via GDS-15 [27]•. NMS are positively correlated with The Parkinson's Disease Questionnaire Summary Index (PDQ-SI), the UPDRS and severity of depression. Note that NMS such as hyposmia, constipation and RBD represent the earliest symptoms of PD [27]•.
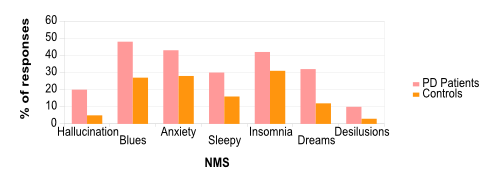
Figure 2. Study of non-motor symptoms in Parkison's Disease patients. .
(Adapted from Chaudhuri et al., 2008.) Results of the NMSQuest with PD patients and controls (same age) showing the percent of people who answer yes to the appearance of the symptoms during their disease.
Genetic Models for Parkinson's Disease
All these symptoms have been observed and measured in PD patients. But the current main problem is the lack of valid models to test treatments and understand how they appear. Indeed, animal models already used recapitulate the different symptoms but are not based on the true origin (for the genetic forms). On the contrary, models based on mutations found in PD patients do not mimic correctly all symptoms (histological, motor and non-motor symptoms). That is why it is necessary to develop genetic models which are able to reproduce the origins of the disease and lead to the same symptoms.
Until now, there are three major genetic models used in PD research: α-synuclein (α-syn), LRRK2 and parkin models (Figure 3). Indeed, the first gene identified as involved in PD was the α-syn gene [28]. More recently, new mutations have been found in Leucine-Rich Repeat Kinase 2 (LRRK2), Parkin, PTEN-Induced Kinase 1 (PINK1) and DJ-1 genes. Theses proteins are associated with mitochondrial metabolism or are involved in protein degradation through the ubiquitine proteasome system [29]•. This review will then recapitulate these three models as the most relevant and the most studied until today.

Figure 3. Transgenic mice generated for PD study. .
Several models have been performed to establish most symptoms (motor and non-motor) of PD. Here are listed the promoters used (blue), the mutations studied and most common (brown), the transgene expression pattern (purple), the damaged structures (pink) and the symptoms observed in mice models (orange).
Models based on autosomic dominant gene mutated
α-synuclein model
The α-syn gene is an autosomal dominant gene. Its overexpression has been developed by using several promoters. Nevertheless, these models do not illustrate all neurological lesions features and behavior of PD patients.
Use of tyrosine hydroxylase promoter (Thp) restricts the α-syn expression to catecholaminergic neurons because tyrosine hydroxylase allows to transform L-tyrosine into L-DOPA, therefore it is specifically expressed in this. Several lines of mice have been generated with this promoter. One of them expresses a construction with the human α-syn protein containing a double mutation A53T plus A30P under Thp in which Richefield and colleagues observed the degeneration of nigrostriatal neurons and a locomotor activity decrease [30]. Other lines have been generated with truncated α-syn under Thp. However it does not lead to degeneration of SN neurons (which occurs at early stages of the disease) but only to nigrostriatal pathology [31]. Chen and Feary also showed that the phosphorylation of Serine 129 of α-syn protein is necessary to increase the pathology in flies [32] and to get a cell loss and a reduced dopamine level in mice even if it is not enough to cause significant neurodegeneration [33].
Other transgenic mice have been generated with Prion promoter (prpP) controlling human α-syn mutated on A53T position. Use of Prion promoter allows to obtain high quantity of the mutated protein in neurons. The A53T mutation found in PD patients seemed to be link to a large field of phenotype and more particularly to a high frequency of dementia and psychiatric impairments [29]•. Paumier and colleagues studied transgenic mice prpP-A53T and observed sensorimotor dysfunction before the onset of early symptoms of PD such as motor problems and cognitive deficiency. At 6 and 12 months, these mice exhibit loss of spatial memory and signs of long-term depression [34]. Another line has also been conceived with prpP but with human α-syn mutated on E46K. This missense mutation is able to cause PD and authors noted that morphology, histology and biochemistry of LB was closer than ones in prpP-A53T mice. It also leads to the formation of neurofibrillary tangles (tau aggregation) [35]. Regarding the behavior of these mice, it is surprising to observe some mice which develop serious motor impairments while others present no significant differences with wild-type mice, maybe due to their intrinsic heterogeneity [35].Two other promoters can be used: PDGFbeta and Thy1, allowing to observe different pattern of expression [36]. Construction with PDGFbeta promoter is associated with reduced level of dopamine in the striatum. This transgenic line of mice allowed to demonstrate a neuroprotective effect of anti-α-syn antibodies in vivo [37]. Thy1 has the advantage to lead to an expression of wild-type human α-syn in high and widespread levels in cortical and subcortical neurons, and more precisely in SN pars compacta avoiding glial, spinal or neuromuscular pathologies [36]. It has been observed that proteinase K resistant inclusions of α-syn, an important symptoms in PD, are present in olfactory bulb, SN and LC, regions involved in earliest stage symptoms of PD [38,39]. So these mice mimic early NMS of PD, such as olfactory deficits and digestion dysfunctions [40]. However, emotional and cognitive symptoms, and more particularly depressive phenotype have not been found yet α-syn mice models, reducing the validity of these models.
LRRK2 model
LRRK2 is strongly involved in both sporadic and familial α-synucleopathies. LRRK2 impairments lead to dysfunction of cytoskeleton dynamics and of the protein degradation system. The most common mutation is G2019S. Li and colleagues designed a transgenic line of mice carrying mutation R1441G, made with a bacterial artificial chromosome clone in which there is the full length LRRK2 gene mutated on arginine 1441. This model is interesting because it recapitulates the main features of PD such as disturbances in movement and a lower dopamine release linked to nigrostriatal dopaminergic axonal projection impairments [41]. So these mice could be a good model for therapeutic improvements. In a more recent study, Bichler and colleagues tried to investigate if NMS precede MS with this transgenic mouse. They did not find symptoms at early stage but only gastrointestinal disorders [42]. So this transgenic mouse could be a good model with the advantage of a progressive age-dependent phenotype motor deficit for therapeutic improvement but the absence of NMS remains an imperfection. More generally, LRRK2 acts like α-syn: enhancing through an overexpression of the gene increase the neuropathology. On the contrary, a knock-out of the gene reduces cell disorders [43].
Model based on autosomic recessive gene mutated
Parkin model
The parkin gene is an autosomal recessive gene. Parkin protein is involved in proteasome system as an E3 ubiquitine ligase. Recently, it has been shown that Parkin was associated with the mitochondrial outside membrane and could be play a role in the up-keeping of mitochondrial integrity [44]. Parkin acts mainly with PINK1 in the same mitochondrial regulation pathway. A dysfunction of this pathway will stop the process of removal of mitochondrial damages which have as consequences an increase of oxydative stress. This condition seems to favor the α-syn aggregation process [45]. The deletion of the third or the seventh exon in mice leads to get knocked-out mice for Parkin protein which show lesions in nigrostriatal circuits (dopaminergic projections) and within the LC (noradrenergic system). Cognitive and locomotor deficiency were observed [46]••. Surprisingly, an overexpression of Parkin also leads to impairments: progressive degeneration of SN, confirming the hypothesis that some parkin mutants may behave as a dominant-negative [43].
Model based on genetic tools
Not all genetic models are obtained through the generation of transgenic line of mice. Overexpression of α-syn is a way to generate PD models. High concentration of the protein allows to obtain significant neurodegeneration and NMS or MS and to get it more quickly. Viral vectors as lentiviruses or adeno-associated viruses (AAV) can been used. Decressac and colleagues performed an AAV construct using neuron-specific synapsin-1 promoter. They improve the construction in integrating in it a woodchuck hepatitis virus post-transcriptional regulatory element. The efficacy of this model has been demonstrated by the observations of severe motor impairments and degeneration of dopaminergic neurons compared to controls [47]••. Studies are actually done to test the NMS as depression in Per Svenningsson group by overexpress the α-syn in the SN and the VTA.
Estimation of cognitive and emotional troubles in mice models
Behavioral tests on an interesting mice model
An interesting study has been done on an animal model displaying reduced for vesicular storage of monoamine because it only expresses 5% of normal vesicular monoamine transporter 2 (VMAT2-deficient). It has been show that these mice suffer from lots of the NMS [48]•. Through the Forced swim test (FST) and the Tail Suspension Test (TST), measuring mood and depress states of mice by the immobility time, Taylor and colleagues showed that, in early stages (4-6 months), there were no significant difference between normal and VMAT2-deficient mice. But later, at 12-15 months, the immobility time for VMAT2-deficient mice was higher. Moreover, when both VMAT2-deficient mice were exposed to desipramine (an anti-depressive drug), the immobility time of VMAT2-deficient mice decreased to control mice level. It demonstrates that this model could be valid to test the different NMS of PD. On the contrary, the resperine, an inhibitor of VMAT2, accelerated the depressive-like symptoms. Concerning the sleep disturbances, there is no significant difference between controls and VMAT2-deficient mice. Indeed, authors have only noticed a trouble increasing with age, but not accentuated by aging. So VMAT2-deficient-mice seem to be a good model for the study of monoaminergic deficits manifested in human PD.
Another way to develop good mice model is to combine several mutations. For example, Hennis and colleagues described a genetic model with two knock-out: Parkin and DJ-1 (Parkin-/-DJ-1-/-). DJ-1 is a protein involved in the mitochondrial pathway and which participates to the decrease of oxydative stress. Animal models expressing only mutated parkin did not shown NMS. So different tests have been performed in order to test both MS and NMS. Indeed, it has been shown that the locomotor activity and coordination of movements was reduced in Parkin-/-DJ-1-/- mice, more than Parkin-/- alone. In addition, these mice displayed troubles in their reflexes and sensitive mechanisms. The authors tried to understand if the results of the coordination test, which consists in measuring latency for falling for mice from a rotated platform, were due to the fear or the anxiety of the fall. So they performed a FST and observed a decrease of immobility time when anti-depressive drug were administrated to these mice. This model shows depressive states of PD mice. This aspect seems interesting to develop with more tests in order to create more models allowing to study NMS of PD [49].
Others behavioral tests are possible to test the different NMS, like “sucrose preference” or Intracranial self-stimulation (ICSS). Indeed, studies performed in line mouse KO for α-syn gene show that they had superior level of ICSS in comparison with controls. This deletion may lead to an increase of DA release in the mesolimbic terminals [50]. We can also perform open field test and Elevated Plus-Maze (EPM) which are others ways to evaluate a depressive-phenotype in mice.
Conclusion
Many NMS have been listed in scientific literature, but the ones taken for cognitive and emotional are mainly depression, apathy, sleep disorders, emotion troubles like fear, anxiety, and others less detailed here, like hallucinations. All these symptoms are measured with different scales in PD patients, but also used in other pathologies. It could be useful to realize more test as the NMSQuest in order to diagnose earlier PD patients, to the possible extent. Indeed, if this kind of test was more automatic, and if complementary exams were performed as IRM (view of dopaminergic neurons), patients could be cared before a more severe stage of the disease. After that, PD patients who had developed the NMS and MS could be genotyped in order to identify a mutated gene which may be correlated to the different NMS observed before. This approach could serve the development of new models based on these mutated genes discovered, which group together the NMS, combined with known mutation of proteins such as α-synuclein, Parkin, LRRK2, etc. Indeed, after recapitulating of the main genetic models, a huge absence of NMS appears very clear. Few models already developed, like prpA53T, display cognitive troubles as dementia and depression. However it is hard to find one which presents sleep disorders, emotional troubles, apathy, etc., all together. These conclusions could let us think that the generation of one unique model with all PD symptoms is really ambitious, because of the variety of mutations each associated with different dysfunctions. Maybe the research of better models should target the development of different models expressing one cluster of symptoms in order to study this more specifically. The identification of new genes related to NMS could allow to understand which lesions in brain lead to these symptoms. Another point often seen in generation of model, is to take a new pharmacological dimension in using drugs which mimic the different NMS (for example the resperine leads to memory troubles but does not affect motor activity) [51]. But this way makes harder the research of the origins of the symptoms, and only allows trials of new treatments.
Acknowledgments
I thank Dr Per Svenningsson and Dr Dorian Caudal from the Karolinska Institute for their help and their advices concerning this review and Thibaut Deschamps who followed the progression since the beginning, without forget Lucie, Dorian and Dylan who supported me.
References and recommended reading
Papers of particular interest have been highlighted as:
● of special interest
●● of outstanding interest
1. Riederer P, Wuketich S: Time course of nigrostriatal degeneration in parkinson's disease. A detailed study of influential factors in human brain amine analysis. J. Neural Transm. 1976, 38:277–301. [PubMed]
2.
Becker G, Müller A, Braune S, Büttner T, Benecke R, Greulich W, Klein
W, Mark G, Rieke J, Thümler R: Early diagnosis of Parkinson's disease. J.
Neurol. 2002,
249 Suppl 3:III/40–48. [Full Paper][PubMed] ● The authors explain that
the NMS are directly linked to damages of dopaminergic neurons but also
to others structures which belong to the same circuit.
3. Varanese S, Perfetti B, Ghilardi MF, Di Rocco A: Apathy, but not depression, reflects inefficient cognitive strategies in Parkinson's disease [Internet]. PLoS One 2011, 6. [Full Paper][PubMed]
4. Goerlich-Dobre KS, Probst C, Winter L, Witt K, Deuschl G, Möller B, van Eimeren T: Alexithymia—an independent risk factor for impulsive-compulsive disorders in Parkinson's disease. Movement Disorders 2013, doi:10.1002/mds.25679. [Full Paper]
5. Iranzo A, Molinuevo JL, Santamaría J, Serradell M, Martí MJ, Valldeoriola F, Tolosa E: Rapid-eye-movement sleep behaviour disorder as an early marker for a neurodegenerative disorder: a descriptive study. Lancet Neurol 2006, 5:572–577. [Full Paper][PubMed]
6. Schmeichel AM, Buchhalter LC, Low PA, Parisi JE, Boeve BW, Sandroni P, Benarroch EE: Mesopontine cholinergic neuron involvement in Lewy body dementia and multiple system atrophy. Neurology 2008, 70:368–373. [Full Paper][PubMed]
7. Aarsland D, Påhlhagen S, Ballard CG, Ehrt U, Svenningsson P: Depression in Parkinson disease--epidemiology, mechanisms and management. Nat Rev Neurol 2012, 8:35–47. [Full Paper][PubMed]
8.
Grinberg LT, Rueb U, Alho AT di L, Heinsen H: Brainstem pathology and non-motor symptoms in
PD. J. Neurol. Sci. 2010, 289:81–88. [Full Paper][PubMed] ● The authors research the link between the NMS and the MS
through the anatomical
lesions found in the brain of PD patients.
9. Merschdorf U, Berg D, Csoti I, Fornadi F, Merz B, Naumann M, Becker G, Supprian T: Psychopathological symptoms of depression in Parkinson's disease compared to major depression. Psychopathology 2003, 36:221–225. [Full Paper][PubMed]
10. Yamamoto M, Pohli S, Durany N, Ozawa H, Saito T, Boissl KW, Zöchling R, Riederer P, Böning J, Götz ME: Increased levels of calcium-sensitive adenylyl cyclase subtypes in the limbic system of alcoholics: evidence for a specific role of cAMP signaling in the human addictive brain. Brain Res. 2001, 895:233–237. [PubMed]
11. Schintu N, Zhang X, Svenningsson P: Studies of depression-related states in animal models of Parkinsonism. J Parkinsons Dis 2012, 2:87–106. [Full Paper][PubMed]
12. Kasten M, Klein C: The many faces of alpha-synuclein mutations. Mov. Disord. 2013, 28:697–701. [Full Paper][PubMed]
13. Montgomery EB Jr, Koller WC, LaMantia TJ, Newman MC, Swanson-Hyland E, Kaszniak AW, Lyons K: Early detection of probable idiopathic Parkinson's disease: I. Development of a diagnostic test battery. Mov. Disord. 2000, 15:467–473. [PubMed]
14.
Pluck GC, Brown RG: Apathy in Parkinson's disease.
J. Neurol. Neurosurg.
Psychiatr. 2002,
73:636–642. [PubMed] ● The authors evaluate the
probability to display apathy in association with PD. They observed
indeed that PD patients presented a higher rate of apathy symptom, which
could be liked to cognitive impairments.
15. Tessitore A, Hariri AR, Fera F, Smith WG, Chase TN, Hyde TM, Weinberger DR, Mattay VS: Dopamine modulates the response of the human amygdala: a study in Parkinson's disease. J. Neurosci. 2002, 22:9099–9103. [PubMed]
16. Sprengelmeyer R, Young AW, Mahn K, Schroeder U, Woitalla D, Büttner T, Kuhn W, Przuntek H: Facial expression recognition in people with medicated and unmedicated Parkinson's disease. Neuropsychologia 2003, 41:1047–1057. [PubMed]
17. Delaveau P, Salgado-Pineda P, Witjas T, Micallef-Roll J, Fakra E, Azulay J-P, Blin O: Dopaminergic modulation of amygdala activity during emotion recognition in patients with Parkinson disease. J Clin Psychopharmacol 2009, 29:548–554. [Full Paper][PubMed]
18. Bowers D, Miller K, Bosch W, Gokcay D, Pedraza O, Springer U, Okun M: Faces of emotion in Parkinsons disease: micro-expressivity and bradykinesia during voluntary facial expressions. J Int Neuropsychol Soc 2006, 12:765–773. [Full Paper][PubMed]
19. Miller KM, Okun MS, Marsiske M, Fennell EB, Bowers D: Startle reflex hyporeactivity in Parkinson's disease: an emotion-specific or arousal-modulated deficit?Neuropsychologia 2009, 47:1917–1927. [Full Paper][PubMed]
20. Dietz J, Bradley MM, Okun MS, Bowers D: Emotion and ocular responses in Parkinson's Disease. Neuropsychologia 2011, 49:3247–3253. [Full Paper][PubMed]
21. Yoshida H, Yamada T, Matsuzaki H: [Reflexive and voluntary saccades in Parkinson's disease]. Nippon Ganka Gakkai Zasshi 2002, 106:281–286. [PubMed]
22. Briand KA, Strallow D, Hening W, Poizner H, Sereno AB: Control of voluntary and reflexive saccades in Parkinson's disease. Exp Brain Res 1999, 129:38–48. [PubMed]
23. Clark US, Neargarder S, Cronin-Golomb A: Visual exploration of emotional facial expressions in Parkinson's disease. Neuropsychologia 2010, 48:1901–1913. [Full Paper][PubMed]
24.
Jellinger KA: Synuclein
deposition and non-motor symptoms in Parkinson disease. J.
Neurol. Sci. 2011,
310:107–111. [Full Paper][PubMed] ●● Regarding the damaged structures,
the authors correlated LB, composed of α-synuclein aggregated with NMS. The
lesions involved in sleep troubles remains uncertain but it may be due to
impairments of monoaminergic system.
25.
Chaudhuri KR, Martinez-Martin P, Schapira AHV, Stocchi F, Sethi K,
Odin P, Brown RG, Koller W, Barone P, MacPhee G, et al.: International multicenter pilot study of the first
comprehensive self-completed nonmotor symptoms questionnaire for Parkinson's
disease: the NMSQuest study.
Mov. Disord. 2006, 21:916–923. [Full Paper][PubMed] ●● The authors, after having listed
every NMS which appear in early stages of PD were interested in developing
ways to measure them in order to improve quality life of patients. The
NMSQuest is a promising scale because it allows to evaluate every NMS known
and to diagnose patients before too many neurons would degenerate.
26. Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P, Poewe W, Sampaio C, Stern MB, Dodel R, et al.: Movement Disorder Society-sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov. Disord. 2008, 23:2129–2170. [Full Paper][PubMed]
27.
Duncan GW, Khoo TK, Yarnall AJ, O'Brien JT, Coleman SY, Brooks DJ,
Barker RA, Burn DJ: Health-related
quality of life in early Parkinson's disease: The impact of nonmotor
symptoms. Mov. Disord. 2013, doi:10.1002/mds.25664. [Full Paper][PubMed] ● Duncan and colleagues have compared the different scales performed for
evaluating NMS symptoms like the NMSQuest. They study their sensitivity
to detects the cognitive and emotional impairments, and their
reproducibility.
.
28. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al.: Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997, 276:2045–2047. [PubMed]
29.
Corti O, Lesage S, Brice A: What genetics tells us about the causes and mechanisms of
Parkinson's disease.
Physiol.
Rev. 2011, 91:1161–1218. [Full Paper][PubMed] ● This review summarize the different mutations found
in inherited PD, linked them to neurological lesions which are
also observed in sporadic PD. It could be a beginning to performed
models which would be equivalent to sporadic form of
PD.
30. Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, Wuertzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ: Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp. Neurol. 2002, 175:35–48. [Full Paper][PubMed]
31. Tofaris GK, Garcia Reitböck P, Humby T, Lambourne SL, O'Connell M, Ghetti B, Gossage H, Emson PC, Wilkinson LS, Goedert M, et al.: Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J. Neurosci. 2006, 26:3942–3950. [Full Paper][PubMed]
32. Chen L, Feany MB: Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8:657–663. [Full Paper][PubMed]
33. Wakamatsu M, Ishii A, Iwata S, Sakagami J, Ukai Y, Ono M, Kanbe D, Muramatsu S, Kobayashi K, Iwatsubo T, et al.: Selective loss of nigral dopamine neurons induced by overexpression of truncated human alpha-synuclein in mice. Neurobiol. Aging 2008, 29:574–585. [Full Paper][PubMed]
34. Paumier KL, Sukoff Rizzo SJ, Berger Z, Chen Y, Gonzales C, Kaftan E, Li L, Lotarski S, Monaghan M, Shen W, et al.: Behavioral characterization of A53T mice reveals early and late stage deficits related to Parkinson's disease. PLoS ONE 2013, 8:e70274. [Full Paper][PubMed]
35. Emmer KL, Waxman EA, Covy JP, Giasson BI: E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J. Biol. Chem. 2011, 286:35104–35118. [Full Paper][PubMed]
36. Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E: Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J. Neurosci. Res. 2002, 68:568–578. [Full Paper][PubMed]
37. Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, et al.: Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron 2005, 46:857–868. [Full Paper][PubMed]
38. Fernagut PO, Hutson CB, Fleming SM, Tetreaut NA, Salcedo J, Masliah E, Chesselet MF: Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression. Synapse 2007, 61:991–1001. [Full Paper][PubMed]
39. Halliday GM, Del Tredici K, Braak H: Critical appraisal of brain pathology staging related to presymptomatic and symptomatic cases of sporadic Parkinson's disease. J. Neural Transm. Suppl. 2006, [no volume]. [PubMed]
40. Fleming SM, Tetreault NA, Mulligan CK, Hutson CB, Masliah E, Chesselet M-F: Olfactory deficits in mice overexpressing human wildtype alpha-synuclein. Eur. J. Neurosci. 2008, 28:247–256. [Full Paper][PubMed]
41. Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, et al.: Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat. Neurosci. 2009, 12:826–828. [Full Paper][PubMed]
42. Bichler Z, Lim HC, Zeng L, Tan EK: Non-motor and motor features in LRRK2 transgenic mice. PLoS ONE 2013, 8:e70249. [Full Paper][PubMed]
43. Baptista MAS, Dave KD, Sheth NP, De Silva SN, Carlson KM, Aziz YN, Fiske BK, Sherer TB, Frasier MA: A strategy for the generation, characterization and distribution of animal models by The Michael J. Fox Foundation for Parkinson's Research. Dis Model Mech 2013, 6:1316–1324. [Full Paper][PubMed]
44. Youle RJ, Narendra DP: Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12:9–14. [Full Paper][PubMed]
45. Arduíno DM, Esteves AR, Cortes L, Silva DF, Patel B, Grazina M, Swerdlow RH, Oliveira CR, Cardoso SM: Mitochondrial metabolism in Parkinson's disease impairs quality control autophagy by hampering microtubule-dependent traffic. Hum. Mol. Genet. 2012, 21:4680–4702. [Full Paper][PubMed]
46.
Fleming SM, Fernagut P-O, Chesselet M-F: Genetic mouse models of parkinsonism: strengths and
limitations.
NeuroRx 2005, 2:495–503. [PubMed] ●● The authors show the complexity of the establishment of one unique
model. For example in the KO Parkin model mice present different phenotype. To explain it,
they put forward a hypothesis of environmental and intrinsic factors
which could have an effect
on the appearance or not of the symptoms.
47.
Decressac M, Mattsson B, Lundblad M, Weikop P, Björklund A:
Progressive neurodegenerative and
behavioural changes induced by AAV-mediated overexpression of α-synuclein in
midbrain dopamine neurons.
Neurobiol.
Dis. 2012, 45:939–953.
[Full Paper][PubMed] ●● The authors have generated a rat model not based on
mutation identified in inherited PD but on overexpression of
α-synuclein. To do that they used viral vector
(AAV) and they improve it in using different reponse element in order to
increase the level of α-synuclein in
dopaminergic neurons, and therefore the
degenerescence of these neurons.
48.
Taylor TN, Caudle WM, Shepherd KR, Noorian A, Jackson CR, Iuvone PM,
Weinshenker D, Greene JG, Miller GW: Nonmotor symptoms of Parkinson's disease revealed in an
animal model with reduced monoamine storage capacity. J.
Neurosci. 2009,
29:8103–8113. [Full Paper][PubMed] ● Though the generation of a
transgenic mice deficient for VMAT-2 the authors investigate the NMS found
in PD patients. So this model does not allow to study origin of PD, but is a
way to test treatments.
49. Hennis MR, Seamans KW, Marvin MA, Casey BH, Goldberg MS: Behavioral and neurotransmitter abnormalities in mice deficient for Parkin, DJ-1 and Superoxide Dismutase. PLoS ONE 2013, 8:e84894. [Full Paper][PubMed]
50. Oksman M, Tanila H, Yavich L: Brain reward in the absence of alpha-synuclein. Neuroreport 2006, 17:1191–1194. [Full Paper][PubMed]
51. Souza RR, França SL, Bessa MM, Takahashi RN: The usefulness of olfactory fear conditioning for the study of early emotional and cognitive impairment in reserpine model. Behavioural Processes 2013, 100:67–73. [Full Paper]
