

The involvement of ubiquitin and ubiquitin-like proteins in host defenses against Herpesviruses and the mechanisms by which viruses evade them.
2014/01/21
Abstract
The entry of Herpesviruses in cells to replicate triggers host defense systems. Constitutively expressed proteins contribute to the intrinsic immunity. They constitute nuclear domain 10 by their post-translational modification by the addition of small ubiquitin-like modifier. These domains repress viral gene expression. Moreover, ubiquitin addition on viral proteins leads to their degradation. However, viruses hijack cellular deubiquitinase to prevent this process. Finally, innate immunity is activated by a cascade of ubiquitination which can be counteracted by viral proteins. This review gives an overview of ubiquitin and ubiquitin-like modifications roles in host defenses and the way Herpesviruses counteract these mechanisms.
Table of Contents
Introduction
Herpesviruses infect vertebrate as well as non-vertebrate animals. Eight species of Herpesviruses infect humans. Most of them lead to benign infections, like Herpes Simplex Virus type 1 (HSV-1) causing cold sore near the mouth, but they can be responsible for severe diseases in immunocompromised individuals, like cancers Burkitt lymphoma, caused by Epstein-Barr Virus, EBV [1]. They are enveloped viruses with a genome containing a double strand DNA of 125 to 230 kb allowing them to encode for 80 to 180 viral proteins. This family includes three subfamilies of viruses: Alpha-, Beta- and Gammaherpesviruses [2]. After the infection of a new host, the virus replicates in the infected cells: it is the primary infection. Then the virus is able to enter in a latent phase in wich the viral DNA is maintained in the cells but no virion are produced. This phase is interrupted by reactivation phases during which viral particles are produced. They allow the dispersal of the virus in a new host. The severity of the symptoms during the reactivations is extremely variable [3,4]. As Herpesvirus are able to establish a permanent infection in their hosts, they can evade the host's defense mechanisms established by the immune system.
The immune system is divided in three parts: the intrinsic, the innate and the adaptative systems. The first one is characterized by the presence of intracellular constitutive proteins with antiviral functions that inhibit viral gene expression and viral replication. The second one is induced by viruses outside or in a cell: cellular genes implicated in defenses are induced to protect the cell and surrounding cells. The sensing of viruses triggers signaling pathways leading to antiviral gene expression and protein syntheses, including InterFeroNs (IFNs), IFN Stimulated Genes (ISGs) and Tumor Necrosis Factor (TNF). Repressions made by these two systems can be regulated by post-translational modifications, like the covalent addition of a ubiquitin or a ubiquitin-like protein. Small Ubiquitin-like MOdifier (SUMO) is the main ubiquitin-like protein and possesses 3 isoforms; others are: ISG15, NEDD8 (Neddylin), ubiquitin D (also called FAT10), AuTophaGy-related protein 12 (ATG12) [5]••. Unlike the others, the adaptative immune system requires specialized cells (T lymphocytes, B lymphocytes...) and is longer to be established (several days).
Herpesviruses are the most studied viruses as regards ubiquitin and ubiquitin-like modifications. Here, an overview of recent findings about the roles of those modifications in intrinsic and innate immunity involved in cellular defense against Herpesvirus and ways by which viruses evade this defense is provided.
Modifications by ubiquitin and ubiquitin-like proteins in intrinsic immunity and evasion by Herpesviruses
See Box 1 for details about modifications by ubiquitin and ubiquitin-like proteins.
Sumoylation is involved in inhibition of viral gene expression
In cells newly infected with HSV‑1, mostly during the primary infection, an accumulation of subnuclear globular discrete structures localized near the sites of the viral genome after entry into the nucleus is observed [6]. These structures, found associated with the nuclear matrix, are called ND10 (Nuclear Domain 10) – but also PML NBs (ProMyelocytic Leukemia Nuclear Bodies) or PML Oncogenic Domains (PODs). Major ND10 components are PML, Sp100 (Speckled protein of 100 kDa) and hDaxx (human death domain-associated protein 6) [7]. It has been shown that these 3 components act cooperatively for the restriction of the viral genome [8]. ND10 have a number of different roles within the cell, among them DNA damage response, stress response, apoptosis, senescence, IFN response and viral resistance [5]••.
The accumulation of ND10 previously observed contributes to the intrinsic cellular defense against viral infections. Indeed, ND10 inhibit expression of early genes necessary for the viral replication. Sp100 is one of protein responsible of this inhibition. For instance, during HSV‑1 infection, Sp100 represses the Immediate Early (IE) and early classes proteins expression (Infected Cell Protein 4 – ICP4 - and UL42) and during EBV infection, it regulates viral gene expression [9]. In cells infected by Human CytoMegaloVirus (HCMV), Sp100 restricts the acetylation of histones in viral promoter Major Immediate Early (MIE), resulting in the inhibition of viral gene expression [10]. Sp100 is able to interact with Heterochromatin Protein 1 (HP1), leading to formation of heterochromatin and thus to inhibition of viral DNA transcription [11]. Moreover, hDaxx protein is also required for the repression of viral gene expression, using another strategy: its interaction with Alpha Thalassemia/mental Retardation syndrome X-linked (ATRX) – chromatin remodeling factor belonging to ND10 – leads to chromatin remodeling and repression of viral gene expression [12]•.
The PML protein is the major component of ND10. Its N‑terminal domain contains a RING finger domain, 2 B‑boxes - zinc binding and stabilized domains - and a coiled-coil domain [13]. Those domains constitute a TRIpartite Motif (TRIM), hence the other name of PML: TRIM19. PML possesses 7 isoforms sharing a homologous N‑terminal end, generated by alternative splicing and found in different subnuclear structures [7]. Post-translational modifications of PML are required for its function. They include sumoylation in 3 sites: one in the RING finger (K65), another in the B-box 1 (K160) and the last one at the level of the Nuclear Localization Signal NLS (K490) [7,13]. Finally, a SUMO Interaction Motif (SIM), motif allowing a non-covalent binding with SUMO, is found on PML (from amino acids (aa) 556 to 562) [14]. SUMO modifications, TRIM and SIM are all required for the restriction of HSV-1 infection [14].
PML is required for the formation and maintenance of ND10 (Figure 1, left part). Depletion of PML in cells causes dispersal of hDaxx and Sp100 within the cells. A model was proposed for the formation of ND10: PML is first post-translationally modified by isoform 1 of SUMO: SUMO-1 [9,15]. Indeed, it is proposed that the RING finger of PML could have a SUMO E3 ligase activity, and could sumoylate itself or other proteins [13]. This would allow its nuclear localization [12]• and recruitment of other sumoylated PML thanks to its SIM [13]. However, it was recently proposed that the SIMs are involved in the regulation of ND10 components but not on ND10 formation [14]. Furthermore, the coiled-coil domain of PML permits its hetero and homo-oligomerization and is crucial for ND10 formation [7]. Then, other sumoylated proteins or those possessing a SIM, like hDaxx, can interact with the lattice of PML previously formed to constitute ND10 formation. Depletion of PML induces the alteration of expression or stabilization of Sp100 and also alters its SUMO modification on K297 [9,15,16]. Thus, ND10 formation is highly dependent on sumoylation. However, this modification is not required for Sp100 recruitment to ND10 [15].
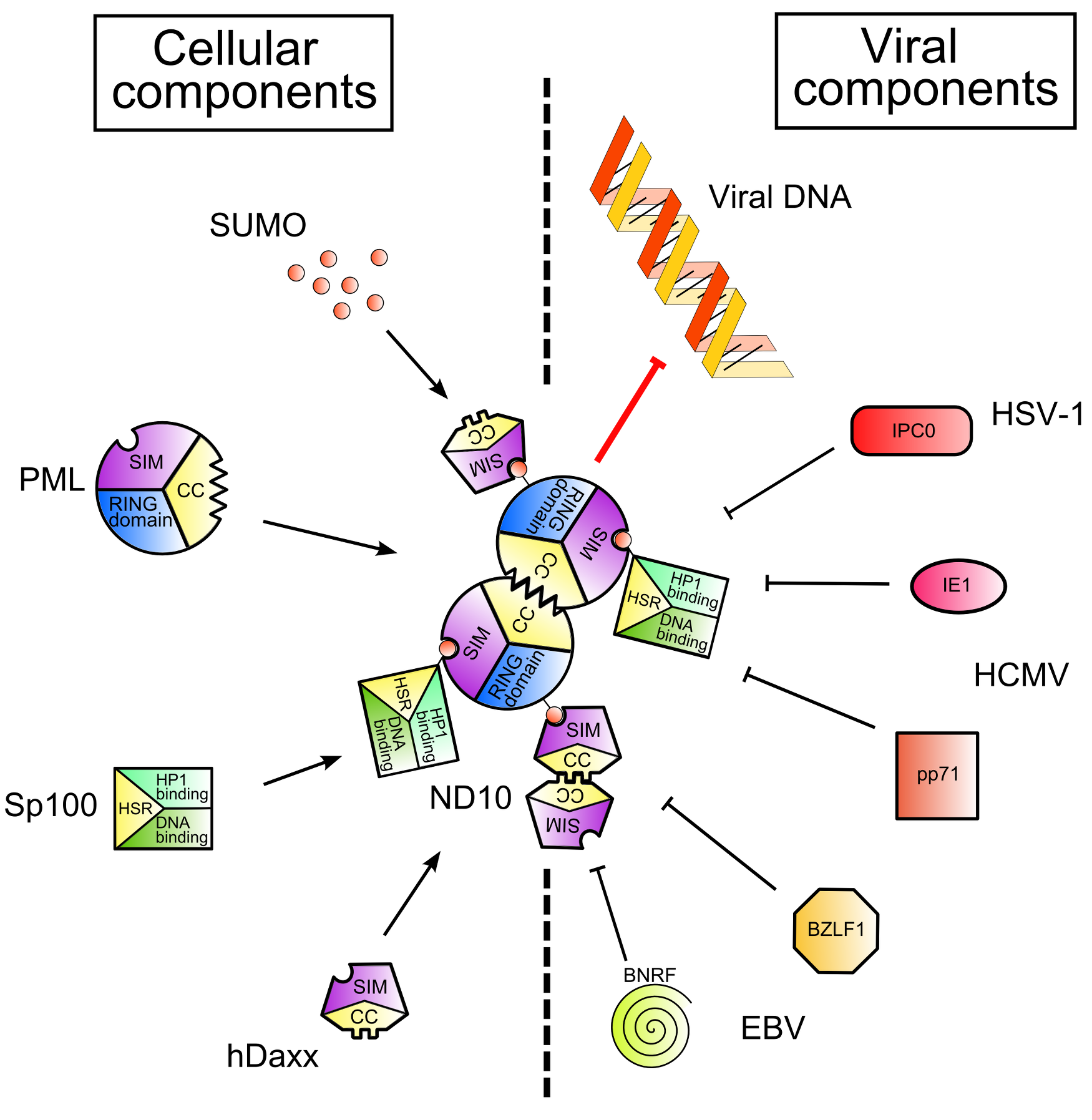
Figure 1. Nuclear Domain 10 (ND10) formation and inhibition by viral proteins. .
Cellular and viral components are shown on the left and on the right of the figure respectively. SUMO (Small Ubiquitin-like MOdifier) proteins post-translationally modify Sp100 and PML, cellular proteins. Thanks to the SIMs – SUMO Interaction Motifs - contained in PML and hDaxx and to the coiled-coil domains of PML and Sp100, ND10 are formed (left part of the figure). This leads to the inhibition of viral gene expression. However, viruses encode proteins which disrupt ND10 and/or degrade ND10 components (right part of the figure), leading to the recovery of viral gene expression.
Viral proteins disrupt ND10, leading to viral gene expression
Whereas the sumoylation of cellular proteins is used to inhibit the viral replication after the entry of the virus in a cell, certain viruses desumoylate these proteins to escape cellular restriction, with or without degrading these proteins themselves (Figure 1, right part). As shown previously, ND10 formation is initiated at sites near viral genome entry in the nucleus and depends on the modification of PML by SUMO-1.
IE1 protein of HCMV, also called IE72, counteracts intrinsic antiviral defense mediated by ND10. This protein is not required for viral replication at high MOI but is required at low MOI [12]. It has been shown to interact with the periphery of ND10 2h post-infection, resulting in the removal of PML, Sp100 and SUMO 4h post-infection [12]. Indeed, IE1 causes the loss of the sumoylated forms of numerous ND10 components in a proteasome independent manner [17]••, resulting in ND10 disruption and inability to repress viral genome expression. IE1 is a hydrophobic (except for C‑terminal region) nuclear phosphoprotein encoded by UL123 [12]. The hydrophobic domain (aa 132 to 346), contrary to the C terminal domain (aa 421 to 491), is required for desumoylation. L174P mutation in IE1 prevents the targeting, desumoylation and disruption of ND10 [18], implying that this residue is essential for the function of IE1. However, the C‑terminal domain is not involved in PML targeting, as it was shown by deletions in IE1 [12]•. Indeed, this domain inhibits IE1 binding on PML [12]•. More recently, it has been shown that IE1 does not possess a SUMO protease activity in vitro against PML but that IE1 induces the desumoylation of PML in vivo [19]. Moreover, in the absence of IE1, the phosphoprotein IE2 (encoded by UL122) is trapped and accumulates in ND10. IE2 is necessary for the progression of the lytic cycle and the expression of delayed-early and late viral genes [20].
EBV also encodes proteins able to disrupt ND10. BamHI Z fragment Leftward open reading Frame 1 protein (BZLF1, also known as Z or Zta or ZEBRA), is an early viral protein required for the activation of viral gene expression. It has been shown that it is also able to disrupt ND10 but this disruption is less efficient when BZLF1 is modified by SUMO-1 [21]. BNRF1 protein interacts with and disrupts the hDaxx/ATRX complex, leading to the activation of the early viral gene expression [22]. Thus, Herpesviruses are able to counteract ND10 defense with the desumoylation and/or the disruption of these structures.
Another strategy used by Herpesviruses is the degradation of cellular proteins implicated in intrinsic immunity, contrary to the basic desumoylation detailed previously. This also occurs when a cell is newly infected.
One of the most studied viral protein possessing this function is ICP0, an IE protein. It interacts with SUMO thanks to its SIM [23]••. It is known for a long time that ICP0 also possesses a RING finger domain which allows the protein to act as an E3 ubiquitin ligase [24]. When the RING finger of ICP0 is mutated, PML and ND10 are not disrupted. This implies that ICP0 is required for ND10 disruption through PML degradation, allowing viral escape from intrinsic defense [5]••[9]. While the degradation of PML happens the fastest, ICP0 also induces the degradation of sumoylated Sp100 in a proteasome dependent way, and more generally, it induces degradation of SUMO conjugates and dispersal of hDaxx and ATRX [23]••. Therefore, ICP0 recognizes its substrates both specifically and via their sumoylation. ICP0 also possesses a number of SIM-Like Sequence (SLS) which is necessary for its functions [23]••.
Other Herpesviruses encode proteins with a similar role to ICP0 of HSV-1. The Murine gamma HerpesVirus 68 (MHV68) tegument protein open reading frame 75c also induces the destruction of PML [8]. Contrary to other Herpesviruses, ORF3 protein of the Herpesvirus Saimiri induces the degradation of Sp100 but not the degradation of PML or hDaxx [25]. Finally, the tegument protein pp71 of HCMV, encoded by the UL82, is also able to interact with ND10, targeting hDaxx and ATRX [26], and then inducing the proteasome-dependent but ubiquitin-independent degradation of hDaxx [12]•[17]••[27].
Viruses use deubiquitinases to restrain the degradation of their proteins
Addition of ubiquitin upon viral proteins by cellular ubiquitin ligases in infected cells leads to the degradation of these viral proteins in a proteasome dependent manner and therefore to a decreased viral replication. This mechanism is included in intrinsic immunity. Furthermore, the sumoylation of viral proteins could also occur in infected cells. IE proteins of HCMV are sumoylated in the host cell and this modification on IE2 inhibits its role of activation of early viral gene expression [28]. However, IE1 sumoylation does not trigger a change in its interaction with ND10 [29]. BZLF1 of EBV and IE1 of Human HerpesVirus 6B (HHV6B) are also immediate early viral proteins sumoylated by the host cell [28]. The accurate function of this sumoylation remains to be clarified.
Viruses may counteract this ubiquitination of their proteins by hijacking ubiquitin proteases. The cellular deubiquitinase Ubiquitin Specific Protease 7 (USP7, also called Herpesvirus Associated Ubiquitin Specific Protease or HAUSP) was the first protein discovered in interaction with ICP0 of HSV-1 [30]•. Indeed, ICP0 induces ubiquitination and degradation of USP7 and also its own degradation by auto-ubiquitination in a RING finger dependent manner [31] (Figure 2). However, interaction of USP7 with ICP0 inhibits this ubiquitination and degradation of ICP0: USP7 stabilizes ICP0 [32]. To determine which action is dominant on the other – degradation of USP7 by ICP0 or stabilization of ICP0 by USP7 -, a study was conducted on mutated ICP0 – which could not bind USP7 - in human fibroblasts. The authors showed that there was a decreasing number of HSV-1 proteins in the cells. They also reduced the level of USP7 proteins with siRNA before the infection with HSV-1 WT (Wild Type) and they observed a lowering rate of ICP0 and IE genes expression. They conclude that stabilization of ICP0 by USP7 is dominant over degradation of USP7 by ICP0 [31]. Other Herpesviruses, like Kaposi Sarcoma HerpesVirus (KSHV), use USP7 to stabilize their proteins [33]. Epstein-Barr Nuclear Antigen 1 (EBNA1) is a protein of EBV required for viral replication and particularly for survival of infected cells during the phase of latency of the virus. USP7 interacts with EBNA1, allowing the removal of polyubiquitin chain in K48, the proteasomal degradation signal (reviewed in [34]). Furthermore, in EBV infected cells, an increase number of USP5, -7, -9, -15i (cellular ubiquitin proteases) has been observed [35]: this may be in response to the rise of cellular stress due to infection.
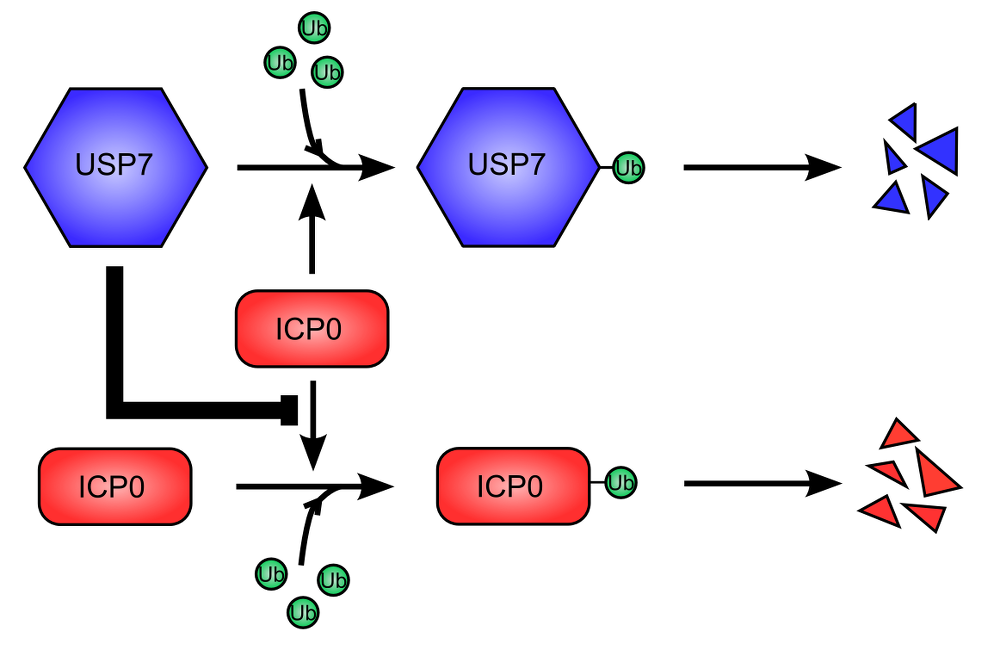
Figure 2. USP7 cellular deubiquitinase is used to stabilize ICP0 viral protein. .
USP7 (Ubiquitin Specific Protease 7) is ubiquitinated, leading to its proteasome-dependent degradation. This is promoted by ICP0 (Infected Cell Protein 0) (top). Similarly, ICP0 auto-ubiquitinylates and is therefore degraded like USP7 (bottom). However, USP7 stabilize ICP0, inhibiting its ubiquitination and degradation. The stabilization of ICP0 is dominant over the degradation of USP7.
Moreover, viruses can also encode deubiquitinases to inhibit the degradation of their proteins. High Molecular Weight Protein (HMWP or pUL48) of HCMV is a ubiquitin protease encoded by the UL48 gene and found in the capsid of HCMV. This protein cleaves only ubiquitin linked with K48. Its active site comprises two important residues: C24 and H162. A mutation in these residues leads to a loss of enzyme activity and slows down virus production [36].
Thus, Herpesvirus have evolved diverse mechanisms to evade the intrinsic immunity set up by the host cells through ubiquitin and ubiquitin-like proteins immediately after the infection of a new cell.
Roles of ubiquitin and ubiquitin-like protein modifications in innate immunity and evasion by Herpesviruses
Viral entry triggers the interferon response
The second line of defense against pathogens is innate immunity. The presence of a pathogen in a cell is sensed by Pattern Recognition Receptors (PRRs) which trigger the activation of signaling pathways, leading to the production of InterFeroNs (IFNs) which set up a local antiviral state (Figure 3). Ubiquitination at different levels of the pathways controls the activation of IFNs production. Herpesvirus, mainly during the phases of latency [3], are sensed by different PRRs located upon membranes or in the cytosol [37]. One sensor able to recognize viral double strand DNA and to induce the downstream pathway to control the virus is Toll-Like Receptor 9 (TLR9), located on endosomal membranes [17]••. TLR9 deficient mice produce less IFNs in response to Murine CytoMegaloVirus (MCMV) infection compared to WT mice, showing the importance of this receptor [38]. TLR9 interaction with viral DNA induces the recruitment of different proteins forming a complex (Figure 3, A). This complex contains in particular the adaptor protein Myeloid Differentiation factor 88 (MyD88) and kinases Interleukin-1 Receptor Associated Kinase (IRAK). Then, this complex recruits TNF Receptor Associated Factor 6 (TRAF6) which oligomerizes, activating its E3 ubiquitin ligase function. Activated TRAF6 polyubiquitinylates itself as well as Receptor-Interacting Protein 1 (RIP1). This leads to the recognition of Nuclear factor κB Essential MOdifier (NEMO, also called Inhibitor of κB Kinase γ: IKKγ). NEMO belongs to IKK complex, so IKK complex is recruited. The consequences of this are the phosphorylation and the ubiquitination of IκB hence the degradation of IκB and the release of NF-κB, which translocates in the nucleus and activates the transcription of IFNβ gene. HCMV is able to activate NF-κB. Furthermore, the first formed complex is also able to recruit TRAF3 (Figure 3, C), promoting its poly-ubiquitination and interaction with IFNs Regulatory Factor 7 (IRF7) to form another complex which acts as a transcription factor, providing IFNs gene transcription (reviewed in [17]••). Herpesvirus are also sensed by TLR3 and TLR7 (sensing of single strand RNA). Although the adaptor protein is different for TLR3 compared to TLR9, the following signaling pathway inducing IFNs production remains similar [17]••.
Interestingly, viral DNA can also be sensed by Retinoic acid Inducible Gene I (RIG-I) directly (HSV-1) or via RNA polymerase III (EBV), Figure 3, B) [37]. The N-terminal domain of RIG-I is K63 ubiquitinated by TRIM25, which allows its interaction with an adaptor called Cardif. Then, TRAF3 is poly-ubiquitinated, leading to the activation of IRF3 – by phosphorylation – and so to IFNs production [39]. It was recently shown that antiviral state induced after infection with HCMV or HSV-1 by the activation of IRF3 is helped by ubiquitination of Stimulator of IFNs Genes (STING) by TRAF56 [40].
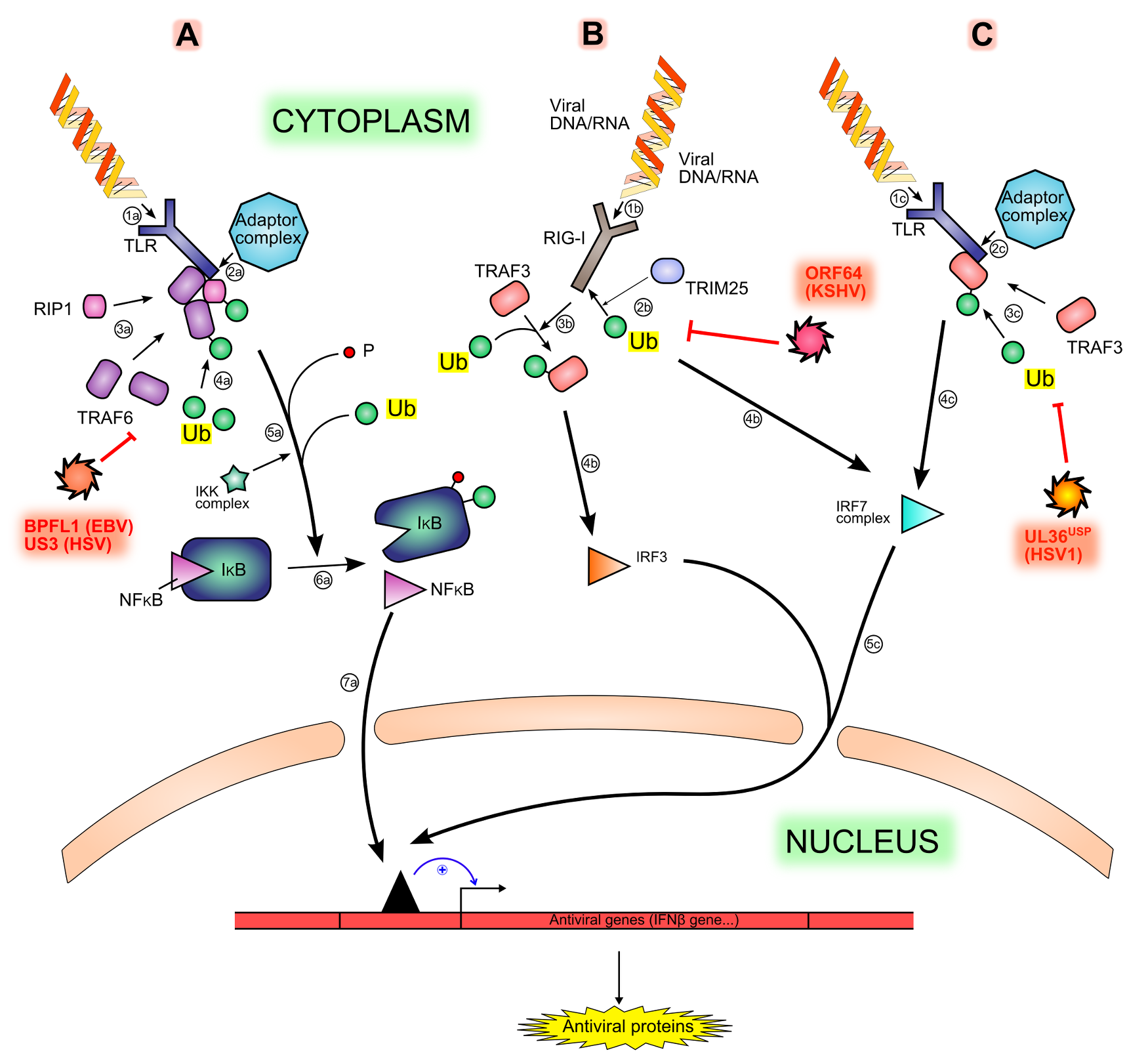
Figure 3. Interferon production signaling pathways and inhibition by viral proteins. .
Viral motifs such as RNA and cytoplasmic unmethylated DNA are sensed by different PRRs (Pattern Recognition Receptors), like TLR (1a, 1b) or RIG-I (1b), triggering different signaling pathways (A, B, C). The sensing is followed by the recruitment of an adaptor complex (2a, 2c), leading to the recruitment and ubiquitination of other proteins (3a, 4a, 3c): TRAF6, RIP1 and TRAF3. Both TRAF6 and RIP1 permit the phosphorylation and then the ubiquitination (5a) of IκB (inhibitor of κB), hence the degradation of IκB and the release of NF-κB (Nuclear Factor κB) (6a). The ubiquitination of TRAF3 leads to the activation of IRF7 (IFNs Regulatory Factor) (4c). Another activation pathway is made by the ubiquitination of the sensor RIG-I by TRIM25 (2b), leading to the activation of IRF3 (3b, 4b) Free NF-κB, activated IRF7 and activated IRF3 translocates in the nucleus and activate cellular antiviral genes expression (7a, 5c). All ubiquitination steps are counteracted by viral deubiquitinases (in red).
It was recently found that NF-κB and IRF3 pathways are dependent on the K27 poly-ubiquination of NEMO by TRIM23. Cells Knock Out (KO) for TRIM23 do not poly-ubiquitinate NEMO, nor have IRF3 and NF-κB responses, contrary to WT cells. These findings show that polyubiquitination of NEMO is essential for antiviral innate response [41]. Thus, IFNs production signalling pathways are highly dependent on ubiquitination of cellular proteins.
The induction of IFNs production in infected cells leads to ISGs production, including ISG15 [28]. Mice defective for production of ISG15 are more susceptible to infection by MHV68 than WT mice [17,42]. This protein leads to the over-expression of IFNβ gene [17]. ISG15 contains two domains ubiquitin-like. It can act as a cytokine and modulate the immune response [17,42]. It can also be linked covalently by its C-terminal domain with other molecules by E1, E2 and E3 specific ligases. Proteins modified by ISG15 are involved in innate immune response: ISGylation stabilizes proteins against degradation. It inhibits IRF3 degradation, hence IFNβ expression and innate immune response increase [17,28]. ISGylation is a reversible process due to proteases, like USP18 [28,42].
Viral deubiquitinases inhibit interferon production
Ubiquitination is an important trigger for innate immune response. However, viruses have developed different strategies to interfere with this ubiquitination (Figure 3). Herpesviruses encode homologous and large tegument proteins, containing N‑terminal DeUBiquitinase (DUB) domains: UL36USP (HSV-1), UL48 (HCMV), M48 (MCMV), BPLF1 (EBV), Open Reading Frame 64 or ORF64 (KSHV and MHV68) [43]••. They are able to cleave K48 and K63 polyubiquitin chains [44]••.
UL36 gene of HSV-1 encodes VP1/2, the largest protein found in its tegument. This protein is required for viral replication. Located at the N‑terminal part of VP1/2, a peptide of 500 amino acids known as UL36USP possesses a DUB activity [45]•. The presence of UL36USP is detected 12h post-infection, after the cleavage of VP1/2 [44]••. UL36USP deubiquitinates TRAF3, hence inhibits the recruitment of a kinase (TBK1: TANK Binding Kinase 1) required for the signaling, and therefore reduces production of IFNβ [44]••. Moreover, UL36USP inhibits TRAF3 dimers formation, required for the activation of IFNs production [44]••. UL36USP C65 mutant does no longer show a DUB activity, implying that this cystein residue is important for the function of this protein, as well as C40 residue [44]••. Deubiquitination of TRAF6 could be performed by the N‑terminal region of BPFL1, an EBV protein, leading to the inhibition of NF-κB signaling pathway during lytic infection and allowing viral replication [46].
Similarly, ORF64 of KSHV inhibits ubiquitination of RIG-I usually performed by TRIM25, leading to the inactivation of the downstream signaling involved in reactivation of lytic infection but not in suppression of NF-κB activation of IFNβ promoter [43]••. ORF64-C29G mutant of KSHV has also no enzymatic activity [43]••, showing the conservation of the cysteine residue among DUBs.
The importance of the cysteine residue for DUB activity is conserved amongst viral DUBs belonging to Herpesviridae family [44]••. HSV encodes for a tegument protein, US3 (Unique Short protein 3), known to inhibit the polyubiquitination of TRAF6. As for EBV, this stops NF-κB signaling pathway and IFNs production [47]•. US3 possesses a kinase activity. It was proposed that the phosphorylation of TRAF6 near the ubiquitination site hides this site and inhibits ubiquitination [47]•.
Herpesviruses hijack cellular deubiquitinases to interfere with interferon production
Viral proteins are able to interact with cellular proteins, leading to the hijacking of their cellular functions. The viral protein ICP0 (HSV-1) interacts with the cellular deubiquitinase USP7 to stop the signalling pathway of IFNs production. USP7 is a protein of 1102 amino acids divided in 3 domains: a TRAF-like domain at the N‑terminal end (aa 58 to 196), a USP domain (aa 208 to 560) and a ICP0 binding domain at the C‑terminal end (aa 622 to 801) [48]. It has a regulatory activity during the signaling of IFNs production: through its TRAF-like and USP7 domains respectively, USP7 binds and deubiquitinylates TRAF6 and IKKγ, resulting in an inhibition of IFNs production. USP7 helps the homeostasis return. However, ICP0 hijacks the regulatory function of USP7 to inhibit IFNs production. Indeed, USP7 is a nuclear protein at rest. Thanks to its NLS, ICP0 enters into the nucleus and thanks to its USP7 binding domain, it binds USP7. This permits the cytoplasmic localization of USP7 and the stimulation of the DUB activity of USP7 [49].
The cellular protein RTA-Associated Ubiquitin Ligase (RAUL) is another cellular protein known to regulate the IFNs production pathways. This protein ubiquitinates IRF3 and IRF7, leading to their degradation by the proteasome. RAUL ubiquitinates itself and is then degraded too (Figure 4). USP7 positively regulates the activity of RAUL by deubiquitination. The RNA Transcriptional Activator (RTA or ORF50) of KSHV recruits USP7 to prevent the destruction of RAUL. Moreover, RTA can increase the activity of RAUL (reviewed in [50]).
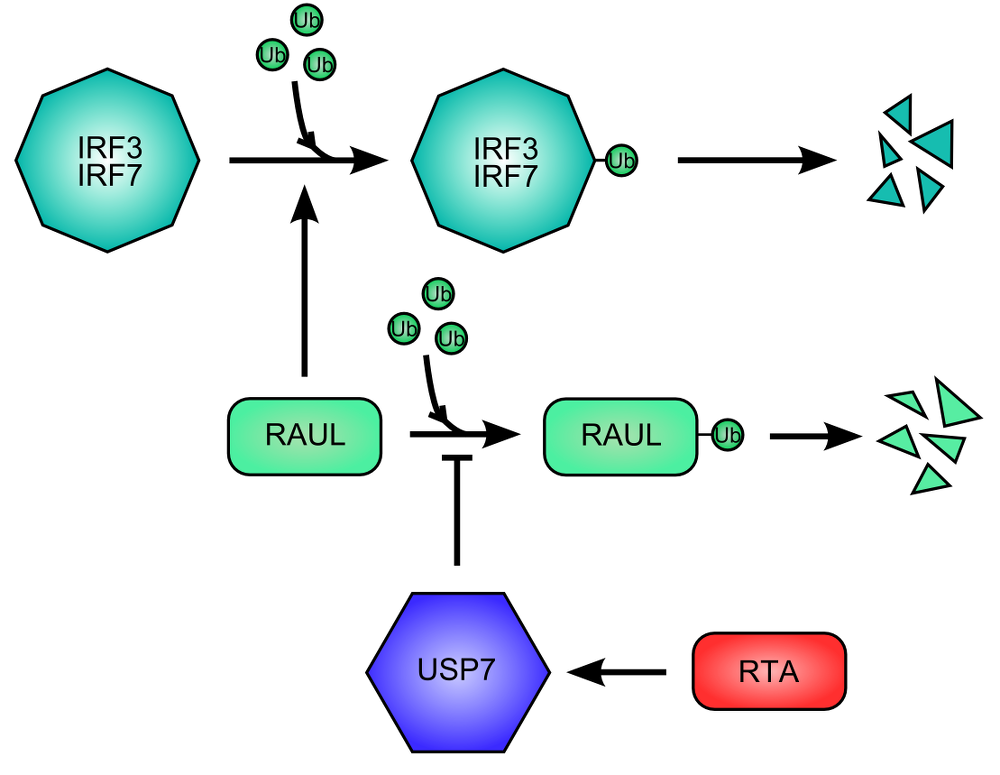
Figure 4. RNA Transcriptional Activator (RTA) protein of Kaposi Sarcoma HerpesVirus promote the destruction of IFN Regulatory Factor 3 and 7. .
The cellular protein RAUL (RTA Associated Ubiquitin Ligase), ubiquitin ligase, promote the degradation of IRF3 end IRF7 by ubiquitination. RAUL auto-ubiquitinates and is then destructed, limiting the degradation of IRF3 and IRF7. The RTA viral activates USP7 for inhibiting the auto-ubiquitination of RAUL.
Conclusion
Post-translational modifications by ubiquitin or ubiquitin-like proteins are used to trigger intrinsic immunity – during the primo-infection and the reactivation phases – and innate immunity – mainly during the latency to control the virus – in infected cells. Recent studies reveal that a lot of viral proteins interact with these modified proteins to avoid viral restriction and to enhance viral replication. Thus, Herpesviruses counteract both intrinsic and innate immunity by removing these modifications or preventing their formation. However, because levels of ubiquitination and sumoylation are low in cells – so difficult to detect -, a lot of studies used for this review are based on over-expression of ubiquitin or ubiquitin-like proteins, which could modify equilibria of enzymatic reactions or more generally the cells' behavior. These methods, even if they are not in vitro studies, are not very close to physiological processes. For future studies, it would be valuable to develop more sensitive makers to detect ubiquitination or other modifications, like antibodies.
Studying interactions between Herpesviruses and hosts cells could provide new targets for antiviral drugs. Thus, inhibitory molecules – not against viral proteins which counteract ubiquitin and ubiquitin-like modifications because of resistance emergence – but against cellular proteins which are hijacked by viruses and not essential for host cells could be new treatments against Herpesviruses.
Acknowledgments
I would like to thank Professor Roger Everett for having welcomed me in his laboratory team and for having suggested this review subject. I also thank Deborah Prevot for her advices and her meticulous readings of this review.
References and recommended reading
Papers of particular interest have been highlighted as:
● of special interest
●● of outstanding interest
1. Carter J, Saunders VA: Virology: Principles and Applications. John Wiley & Sons; 2007.
2. Herpes Simplex Virus - Methods and Protocols. Everett RD: HSV-1 life cycle and biology. Humana Press, 2014.
3. Egan KP, Wu S, Wigdahl B, Jennings SR: Immunological control of herpes simplex virus infections. J. Neurovirol. 2013, 19:328–345. [Full Paper][PubMed][PubMed Central]
4. Grinde B: Herpesviruses: latency and reactivation - viral strategies and host response. J. Oral Microbiol. 2013, 5. [Full Paper][PubMed][PubMed Central]
5. Everett RD, Boutell C, Hale BG:
Interplay between viruses and host
sumoylation pathways.
Nat. Rev.
Microbiol. 2013,
11:400–411. [Full Paper][PubMed] ●● Review giving a complete and precise summary of
SUMO roles in infected cells, for the cells as well as for the
viruses.
6. Lilley CE, Chaurushiya MS, Boutell C, Everett RD, Weitzman MD: The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 2011, 7. [Full Paper][PubMed][PubMed Central]
7. Tavalai N, Stamminger T: Interplay between Herpesvirus infection and host defense by PML nuclear bodies. Viruses 2009, 1:1240–1264. [Full Paper][PubMed][PubMed Central]
8. Glass M, Everett RD: Components of Promyelocytic Leukemia Nuclear Bodies (ND10) act cooperatively to repress herpesvirus infection. J. Virol. 2013, 87:2174–2185. [Full Paper][PubMed][PubMed Central]
9. Everett RD, Parada C, Gripon P, Sirma H, Orr A: Replication of ICP0-null mutant Herpes Simplex Virus type 1 is restricted by both PML and Sp100. J. Virol. 2008, 82:2661–2672. [Full Paper][PubMed][PubMed Central]
10. Kim Y-E, Lee J-H, Kim ET, Shin HJ, Gu SY, Seol HS, Ling PD, Lee CH, Ahn J-H: Human Cytomegalovirus infection causes degradation of Sp100 proteins that suppress viral gene expression. J. Virol. 2011, 85:11928–11937. [Full Paper][PubMed][PubMed Central]
11. Seeler J-S, Marchio A, Sitterlin D, Transy C, Dejean A: Interaction of SP100 with HP1 proteins: A link between the promyelocytic leukemia-associated nuclear bodies and the chromatin compartment. Proc. Natl. Acad. Sci. U. S. A. 1998, 95:7316–7321. [PubMed][PubMed Central]
12. Lee H-R,
Kim D-J, Lee J-M, Choi CY, Ahn B-Y, Hayward GS, Ahn J-H: Ability of the Human Cytomegalovirus IE1 protein to
modulate sumoylation of PML correlates with its functional activities in
transcriptional regulation and infectivity in cultured fibroblast
cells. J. Virol.
2004, 78:6527–6542. [Full Paper][PubMed][PubMed Central] ● This article, although not recent, permits to have
a precise description of IE1 protein of HCMV: roles and domains.
13. Shen TH, Lin H-K, Scaglioni PP, Yung TM, Pandolfi PP: The mechanisms of PML-nuclear body formation. Mol. Cell 2006, 24:331–339. [Full Paper][PubMed][PubMed Central]
14. Cuchet D, Sykes A, Nicolas A, Orr A, Murray J, Sirma H, Heeren J, Bartelt A, Everett RD: PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication. J. Cell Sci. 2011, 124:280–291. [Full Paper][PubMed][PubMed Central]
15. Sternsdorf T, Jensen K, Reich B, Will H: The nuclear dot protein Sp100, characterization of domains necessary for dimerization, subcellular localization, and modification by small Ubiquitin-like modifiers. J. Biol. Chem. 1999, 274:12555–12566. [Full Paper][PubMed]
16. Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A: PML contributes to a cellular mechanism of repression of Herpes simplex Virus type 1 infection that is inactivated by ICP0. J. Virol. 2006, 80:7995–8005. [Full Paper][PubMed][PubMed Central]
17.
Randall RE, Goodbourn S: Interferons and viruses: an interplay between induction,
signalling, antiviral responses and virus countermeasures. J. Gen.
Virol. 2008,
89:1–47. [Full Paper][PubMed] ●● This review explains the diverse IFNs production
signaling pathways
activated when a virus is sensed by a cell.
18. Muller S, Dejean A: Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J. Virol. 1999, 73:5137–5143. [PubMed][PubMed Central]
19. Kang H, Kim ET, Lee H-R, Park J-J, Go YY, Choi CY, Ahn J-H: Inhibition of SUMO-independent PML oligomerization by the human cytomegalovirus IE1 protein. J. Gen. Virol. 2006, 87:2181–2190. [Full Paper][PubMed]
20. Marchini A, Liu H, Zhu H: Human Cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 2001, 75:1870–1878. [Full Paper][PubMed][PubMed Central]
21. Hagemeier SR, Dickerson SJ, Meng Q, Yu X, Mertz JE, Kenney SC: Sumoylation of the Epstein-Barr Virus BZLF1 protein inhibits its transcriptional activity and is regulated by the virus-encoded protein kinase. J. Virol. 2010, 84:4383–4394. [Full Paper][PubMed][PubMed Central]
22. Tsai K, Thikmyanova N, Wojcechowskyj JA, Delecluse H-J, Lieberman PM: EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog. 2011, 7. [Full Paper][PubMed][PubMed Central]
23. Boutell C, Cuchet-Lourenco D,
Vanni E, Orr A, Glass M, McFarlane S, Everett RD: A viral
Ubiquitin ligase has substrate preferential SUMO targeted Ubiquitin ligase activity
that counteracts intrinsic antiviral defence. PLoS
Pathog. 2011, 7. [Full
Paper][PubMed][PubMed Central] ●● Paper finding the mechanisms by which ICP0 interacts with ND10 and
counteracts their antiviral actions. ICP0 induces the proteasome-dependent
degradation of sumoylated proteins. SIMs contained in ICP0 and the specificity
of the target are used for targeting substrates.
24. Hagglund R, Van Sant C, Lopez P, Roizman B: Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. U. S. A. 2002, 99:631–636. [Full Paper][PubMed][PubMed Central]
25. Full F, Reuter N, Zielke K, Stamminger T, Ensser A: Herpesvirus Saimiri antagonizes Nuclear Domain 10-instituted intrinsic immunity via an ORF3-mediated selective degradation of cellular protein Sp100. J. Virol. 2012, 86:3541–3553. [Full Paper][PubMed][PubMed Central]
26. Hwang J, Kalejta RF: Human Cytomegalovirus protein pp71 induces Daxx SUMOylation. J. Virol. 2009, 83:6591–6598. [Full Paper][PubMed][PubMed Central]
27. Lukashchuk V, McFarlane S, Everett RD, Preston CM: Human Cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from Nuclear Domain 10 at early stages of infection. J. Virol. 2008, 82:12543–12554. [Full Paper][PubMed][PubMed Central]
28. Isaacson MK, Ploegh HL: Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 2009, 5:559–570. [Full Paper][PubMed]
29. Gravel A, Dion V, Cloutier N, Gosselin J, Flamand L: Characterization of human herpesvirus 6 variant B immediate-early 1 protein modifications by small ubiquitin-related modifiers. J. Gen. Virol. 2004, 85:1319–1328. [Full Paper][PubMed]
30. Everett
RD, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J: A novel ubiquitin-specific protease is dynamically
associated with the PML nuclear domain and binds to a herpesvirus regulatory
protein.EMBO J.
1997, 16:1519–1530. [Full Paper][PubMed][PubMed Central] ● This is the first paper in which the authors had
discovered a major cellular ubiquitin protease, USP7.
31. Boutell C, Canning M, Orr A, Everett RD: Reciprocal activities between Herpes Simplex Virus type 1 regulatory protein ICP0, a Ubiquitin E3 Ligase, and Ubiquitin-specific protease USP7. J. Virol. 2005, 79:12342–12354. [Full Paper][PubMed][PubMed Central]
32. Canning M, Boutell C, Parkinson J, Everett RD: A RING finger Ubiquitin ligase is protected from autocatalyzed ubiquitination and degradation by binding to Ubiquitin-specific protease USP7. J. Biol. Chem. 2004, 279:38160–38168. [Full Paper][PubMed]
33. Ashizawa A, Higashi C, Masuda K, Ohga R, Taira T, Fujimuro M: The Ubiquitin system and Kaposi's Sarcoma-associated herpesvirus. Front. Microbiol. 2012, 3:66. [Full Paper][PubMed]
34. Frappier L: The Epstein-Barr virus EBNA1 protein. Scientifica (Cairo) 2012, 2012. [Full Paper][PubMed][PubMed Central]
35. Ovaa H, Kessler BM, Rolen U, Galardy PJ, Ploegh HL, Masucci MG: Activity-based ubiquitin-specific protease (USP) profiling of virus-infected and malignant human cells. Proc. Natl. Acad. Sci. U. S. A. 2004, 101:2253–2258. [Full Paper][PubMed][PubMed Central]
36. Wang J, Loveland AN, Kattenhorn LM, Ploegh HL, Gibson W: High-molecular-weight protein (pUL48) of Human Cytomegalovirus Is a competent deubiquitinating protease: mutant viruses altered in its active-site cysteine or histidine are viable. J. Virol. 2006, 80:6003–6012. [Full Paper][PubMed][PubMed Central]
37. Paludan SR, Bowie AG, Horan KA, Fitzgerald KA: Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 2011, 11:143–154. [Full Paper][PubMed] [PMC3686362]
38. Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, et al.: Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. U. S. A. 2004, 101:3516–3521. [Full Paper][PubMed][PubMed Central]
39. Saha SK, Pietras EM, He JQ, Kang JR, Liu S-Y, Oganesyan G, Shahangian A, Zarnegar B, Shiba TL, Wang Y, et al.: Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006, 25:3257–3263. [Full Paper][PubMed][PubMed Central]
40. Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, Kawai T, Akira S: The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 2010, 33:765–776. [Full Paper][PubMed]
41. Arimoto K, Funami K, Saeki Y, Tanaka K, Okawa K, Takeuchi O, Akira S, Murakami Y, Shimotohno K: Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl. Acad. Sci. U. S. A. 2010, 107:15856–15861. [Full Paper][PubMed][PubMed Central]
42. Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, Wolff T, Osiak A, Levine B, Schmidt RE, Garcia-Sastre A, et al.: IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. U. S. A. 2007, 104:1371–1376. [Full Paper][PubMed][PubMed Central]
43. Inn K-S, Lee S-H, Rathbun JY,
Wong L-Y, Toth Z, Machida K, Ou J-HJ, Jung JU: Inhibition of
RIG-I-mediated signaling by Kaposi's sarcoma-associated herpesvirus-encoded
deubiquitinase ORF64. J. Virol. 2011, 85:10899–10904. [Full
Paper][PubMed][PubMed Central] ●● Study clearly showing that RIG-I is a sensor of KSHV and that ORF64,
viral deubiquitinase, can counteract the ubiquitination of RIG-I required for
the activation of the IFNs signaling pathway.
44.
Wang S, Wang K, Li J, Zheng C: Herpes Simplex Virus 1 Ubiquitin-specific protease UL36
inhibits beta interferon production by deubiquitinating
TRAF3. J. Virol.
2013, 87:11851–11860. [Full Paper][PubMed] ●● Paper presenting
UL36USP roles during
infection by Herpesviruses: TRAF3, an important protein of the IFNs
production signaling pathways, is
the substrate of this viral deubiquitinase.
45.
Kattenhorn LM, Korbel GA, Kessler BM, Spooner E, Ploegh HL:
A deubiquitinating enzyme encoded
by HSV-1 belongs to a family of cysteine proteases that is conserved across the
family Herpesviridae.
Mol. Cell 2005, 19:547–557. [Full Paper][PubMed] ● First article which discover the viral deubiquitinase
UL36USP.
46. Saito S, Murata T, Kanda T, Isomura H, Narita Y, Sugimoto A, Kawashima D, Tsurumi T: Epstein-Barr virus deubiquitinase downregulates TRAF6-mediated NF-kB signaling during productive replication. J. Virol. 2013, 87:4060–4070. [Full Paper][PubMed][PubMed Central]
47.
Sen J, Liu X, Roller R, Knipe DM: Herpes simplex virus US3 tegument protein inhibits
Toll-like receptor 2 signaling at or before TRAF6
ubiquitination.
Virology 2013, 439:65–73. [Full Paper][PubMed] ● Paper showing that US3 protein of HSV could reduce the
polyubiquitination of TRAF6, an important protein of the IFNs production
signaling pathway, thanks to its kinase activity.
48. Hu M, Li P, Li M, Li W, Yao T, Wu J-W, Gu W, Cohen RE, Shi Y: Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 2002, 111:1041–1054. [PubMed]
49. Daubeuf S, Singh D, Tan Y, Liu H, Federoff HJ, Bowers WJ, Tolba K: HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 2009, 113:3264–3275. [Full Paper][PubMed]
50. Yu Y, Hayward GS: The ubiquitin E3 ligase RAUL negatively regulates type I interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 2010, 33:863–877. [Full Paper][PubMed][PubMed Central]
51. Randow F, Lehner PJ: Viral avoidance and exploitation of the ubiquitin system. Nat. Cell Biol. 2009, 11:527–534. [Full Paper][PubMed]
